Answer:
3. Homogenous and unsaturated
Explanation:
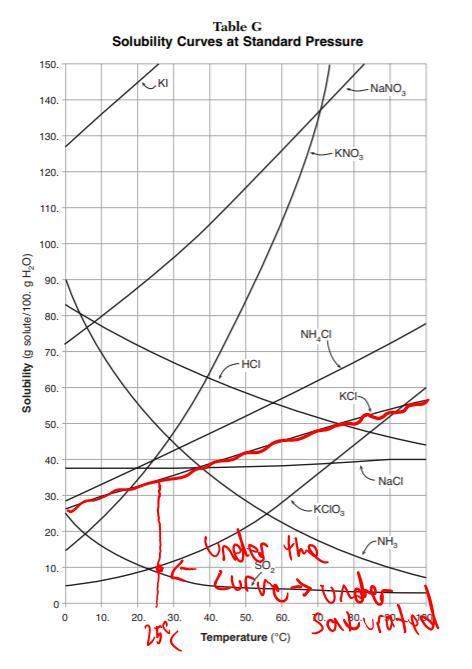
Since your given information mentions 50 grams of solvent and 5 grams of solute, you must double each amount because "
Table G Solubility Curves" in the Regents Chemistry Reference Table notates curves based off of 100 g samples of water. Therefore, you must look for how soluble 10 grams of KCl is in 100 grams of water at 25 degrees Celsius.
As the picture shows, 10 grams of KCl is well below the curve for saturation, meaning that the solution is undersaturated.
As for the second part of the answer, we can tell that the solution is homogenous (of uniform composition) because there is no possibility of the salt, KCl, falling out of solution and making the mixture heterogenous (of mixed composition).
Putting all of those ideas together, we can tell that the answer to this question is:
3. Homogenous and unsaturated
Ledger balance indicates whether right amounts were posted to the correct general ledger accounts